Phylogenetic Reconstruction
Jump to navigation
Jump to search
A Phylogenetic Reconstruction is a reconstruction task of building Phylogenetic Trees.
- Context:
- It can range from being a Parsimony Phylogenetic Reconstruction Task, to being a Distance Phylogenetic Reconstruction Task, to being a Probabilistic Phylogenetic Reconstruction Task.
- See: Taxonomy, Genome, Evolution.
References
2015
- (Shi-Qionghuang et al., 2015) Shi-Qionghuang, Wei Zheng, and Wei-Min Zheng (2015, September). "A brief review to phylogenetic reconstruction by maximum parsimony". In Proccedings of the Fifth International Conference on Instrumentation and Measurement, Computer, Communication and Control (IMCCC 2015) (pp. 830-835). IEEE.
2014
- (De Bruyn et al., 2014) ⇒ Alexandre De Bruyn, Darren P. Martin, and Pierre Lefeuvre (2014). "Phylogenetic reconstruction methods: an overview". In Molecular Plant Taxonomy (pp. 257-277). Humana Press, Totowa, NJ. DOI: /10.1007/978-1-62703-767-9_13.
- QUOTE: Sequence-based phylogenetic reconstructions can be divided into five main steps: (1) choosing a genome region to study, (2) identifying and retrieving sets of homologous sequences from the same genome region of related individuals, (3) aligning homologous nucleotide/amino acid sites within these sequences, (4) constructing a phylogenetic tree, and (5) visualizing the tree. Although one might assume that the fourth step is the most complex, it is very important to realize that to produce a meaningful tree, none of these steps should be neglected.
2007
- (Barton et al., 2007) ⇒ Nicholas H. Barton, Derek E.G. Briggs, Jonathan A. Eisen, David B. Goldstein, Nipam H. Patel (2007) Chapter 27:Phylogenetic Reconstruction In: Evolution, 1-55.
- QUOTE: A phylogenetic tree is the only figure in On the Origin of Species, evidence of the central importance of such trees to evolutionary biology. As discussed in Chapter 5, a phylogenetic tree is a graphical representation of the evolutionary relationships among entities that share a common ancestor. Those entities can be species, genes, genomes, or any other operational taxonomic unit (OTU). More specifically, a phylogenetic tree, with its pattern of branching, represents the descent from a common ancestor into distinct lineages. It is critical to understand that the branching patterns and branch lengths that make up a phylogenetic tree can rarely be observed directly, but rather they must be inferred from other information.
2006
- (Harrison & Langdale, 2006) ⇒ C. Jill Harrison, Jane A. Langdale (2006). "A step by step guide to phylogeny reconstruction". The Plant Journal 45, no. 4 (2006): 561-572. DOI:10.1111/j.1365-313X.2005.02611.x
- QUOTE: A phylogeny is the evolutionary history of a group of entities. Given that this can only truly be known in exceptional circumstances, the main aim of phylogeny reconstruction is to describe evolutionary relationships in terms of relative recency of common ancestry. These relationships are represented as a branching diagram, or tree, with branches joined by nodes and leading to terminals at the tips of the tree (Figure 1).
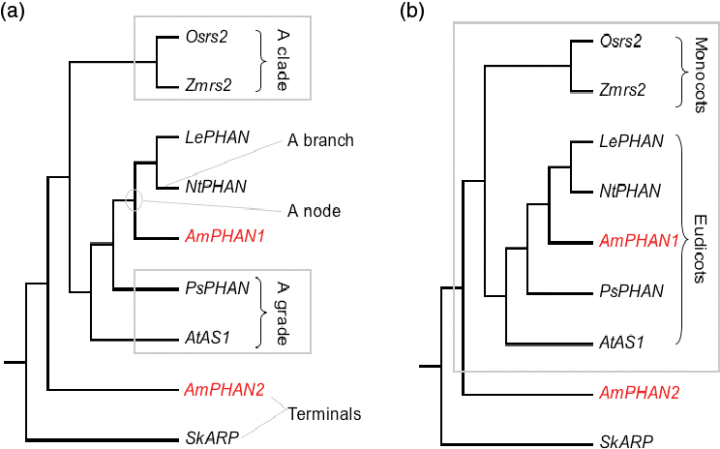
Figure 1 Some phylogenetic terminology illustrated using a phylogeny of ARP genes redrawn from Harrison et al. (2005) and rooted on a Selaginella ARP gene, SkARP.
In the phylogeny, Osrs2 is the sister terminal to Zmrs2, and together these form a monophyletic group. PsPHAN and AtAS1 are paraphyletic with respect to the monphyletic sister group containing AmPHAN, NtPHAN and LePHAN. ‘Clade’ and ‘monophyletic group’ can be used interchangeably, as can ‘grade’ and ‘paraphyletic group’.
- QUOTE: A phylogeny is the evolutionary history of a group of entities. Given that this can only truly be known in exceptional circumstances, the main aim of phylogeny reconstruction is to describe evolutionary relationships in terms of relative recency of common ancestry. These relationships are represented as a branching diagram, or tree, with branches joined by nodes and leading to terminals at the tips of the tree (Figure 1).
- (a) Terminals, branches, nodes, a clade and a grade are indicated.
- (b) To date, Antirrhinum is the only species reported to have two ARP genes, AmPHAN1 and AmPHAN2, and these are paralogues. The box indicates a monophyletic (orthologous) gene group.
2004
- (Isaev) ⇒ Alexander Isaev. (2004). “Introduction to Mathematical Methods in Bioinformatics." Springer. ISBN:3540219730.
- QUOTE: The process of building phylogenetic trees that are in some sense optimal for a given set of OTUs is called phylogenetic reconstruction and involves the following basic steps.
- (i) Choosing a family of homologous sequences as OTUs (...)
- (ii) Putting the sequences into multiple alignment and obtaining a reduced multiple alignment by discarding the columns that contain gaps (...)
- (iii) Inferring a phylogenetic tree from the reduced multiple alignment (...)
- In reality, when the sequences involved are much longer and the number of OTUs is much larger, the resulting reduced multiple alignment is analyzed by a particular method of phylogenetic reconstruction. Such methods can be classified into three groups:
- (a) Parsimony methods,
- (b) Distance methods,
- (c) Probabilistic methods arising from the maximum likelihood approach.